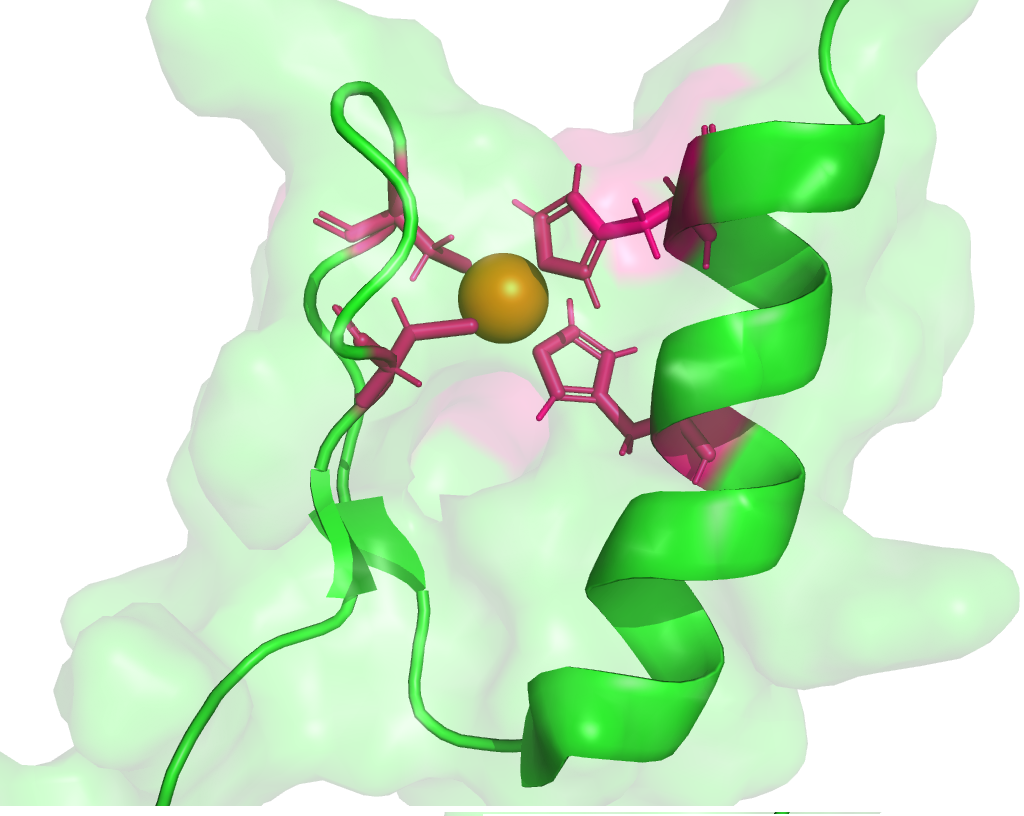
ZnF-Prot: Zinc finger motifs in proteome
Proteome wide automated prediction of zinc finger motifs
Click the above button for Zinc finger motif prediction
Proteome wide automated prediction of zinc finger motifs
Click the above button for Zinc finger motif prediction
The overall flowchart of sequence based zinc finger motif prediction methododlogy
1. Zinc finger motif Pattern search approach
2. Sequence to Pfam HMM profile search approach
3. Choice of e-value threshold for Pfam HMM profile search
4. Prediction of ZnF by comparing pattern search and Pfam HMM profile search approach
5. Prediction of ZnF through pairwise comparison between HMM profile and query
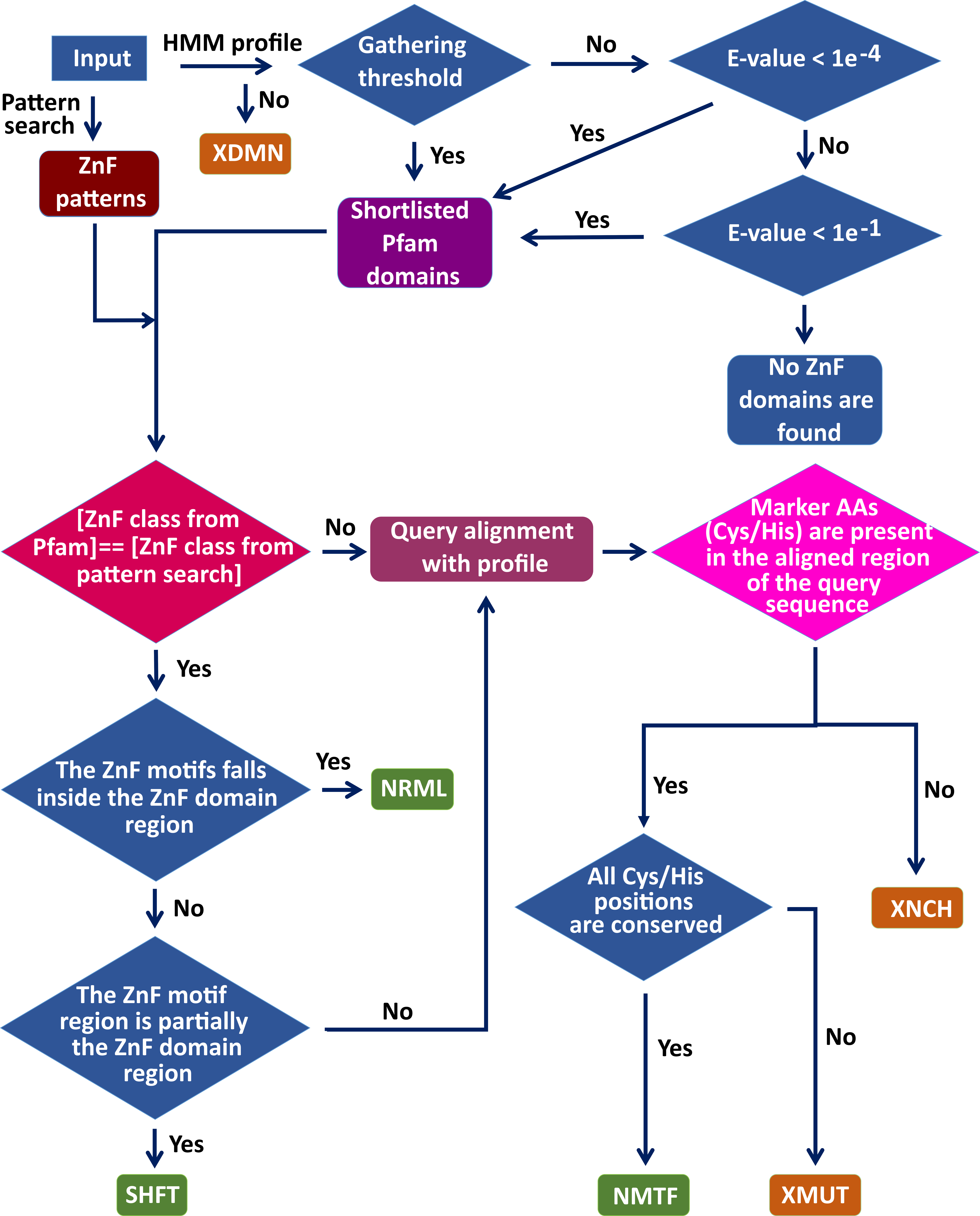
When the query sequence is submitted as an input, the zinc finger motif patterns will be searched against the local database which contains 71 zinc finger motif patterns. After pattern search, the web server will report any possible zinc finger motif patterns which belong to 31 different zinc finger domain classifications. Due to the shorter length of the ZnF sequence patterns, the output from the pattern search approach may contain false positive(s). All the zinc finger motif patterns reported for the query sequence were considered and subjected to further prediction analysis to overcome this issue.
Sequence to HMM profile search approach was used to ignore the false positive results and increase the accuracy of zinc finger domain prediction. The HMMER software is used for the sequence to HMM profile search. Totally 288 Pfam HMM profiles were collected and compiled into a single HMM profile. Further, the hmmpress command was used to convert the HMM profile from HMM database format into a binary format for hmmscan and hmmsearch. The hmmscan command was used to search the query protein sequence against the target HMM profile for the domain search. By considering the gathering threshold cutoff, sequence to Pfam HMM profile search approach will give the significant Pfam families against query sequence.
If no Pfam families are reported through the above criteria, an e-value cutoff of 1e-04 is used to shortlist the Pfam families. Note that the e-value cut-off of 1e-04 was chosen based on the systematic analysis of 610 test cases, wherein, the e-value less than 1e-04 is observed for the successful prediction. To further eliminate the false negative prediction by retaining the accuracy of prediction, in the next round, a relaxation of 1e-01 is given for the e-value cutoff to search for the Pfam family. Note that this relaxation has been incorporated since one of the 610 test cases exhibited the e-value cutoff of 1e-03. Finally, the Pfam families with the e-value >1e-01 have been excluded for the next round of ZnF domain prediction.
The results obtained from the zinc finger motif pattern search approach and sequence to Pfam HMM profile search approach are compared to remove the false positives from both the approaches. If both the approaches report the presence of one or more common zinc finger domains in the query sequence, it is considered as an accurate zinc finger domain prediction.Notably, there are two different types of pattern search and Pfam HMM profile search comparisons are done. The first one is a straight forward method, wherein, the residue numbers of the pattern search and Pfam HMM profile search matches (normal (NRML) in Figure 2). There may be a situation, in which, there is a shift in the residue numbers of the ZnF motif predicted using the pattern search falls partially within the ZnF domain predicted using Pfam HMM profile search. Since this still represent the presence of the ZnF motif, this has been included as an accurate prediction, but with a shift (shift (SHFT)).
However, there may be insertion(s) and/or deletion(s) and/or mutation(s) of amino acids in the finger motif patterns across different organisms due to evolution. While the pattern search may lead to a false negative ZnF prediction for such scenario, the Pfam HMM profile search can still report the presence of ZnF motif. Thus, to reduce the false-negative predictions under such circumstances, ZnF-Prot proceeds with the prediction through Pfam HMM profile-based prediction. In this case, the pairwise sequence alignment of HMM profile to query sequence has been carried out to find out the presence of conserved zinc coordinating residues (cysteine(s) or histidine(s)) in the HMM profile as well as in the query sequence. If both the HMM profile and query sequence have the marker residue(s) (cysteine(s) or histidine(s)) then it is considered that the query sequence has the potential ZnF motif (no motif (NMTF)). Since there is no ZnF pattern is in seen in the query, ZnF simply reports the presence of a ZnF domain without mentioning the position of the motif. However, if there are no conserved marker amino acids found in the query, then, ZnF-Prot reports that the query has the mutated ZnF motif (not considered as ZnF motif due to the presence of mutation (XMUT)). In contrast, if the pairwise aligned HMM profile region doesn’t have the conserved marker amino acids (cysteine(s) or histidine(s)), then ZnF-Prot directly reports the absence of any ZnF motif in the query (no pattern and no conserved C and H (XNCH)).